اسلاید قبلی
اسلاید بعدی
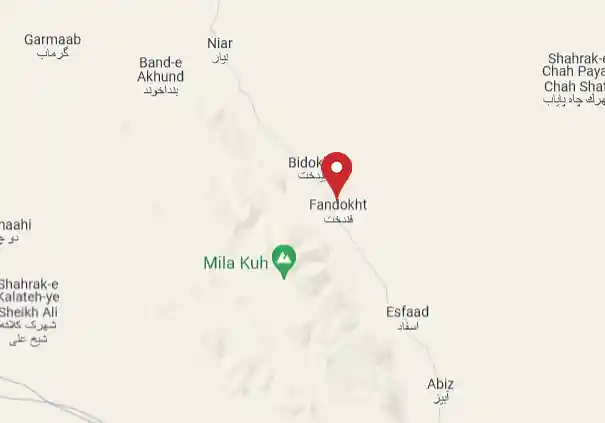
زعفران فندخت محصولی از مزارع زیرکوه قائنات
با زعفران خالص ما، به سفرهی خود چاشنی اضافه کنید. طعمی غیرقابل مقاومت که باعث شادی و نشاط خانواده خواهد شد.









با زعفران خالص ما، به سفرهی خود چاشنی اضافه کنید. طعمی غیرقابل مقاومت که باعث شادی و نشاط خانواده خواهد شد.
زعفران اعلاء قائنات را از روستای فندخت بخواهید.
ما با افتخار اعلام میکنیم که بهعنوان یک تولید کننده و فروشنده معتبر زعفران، به مشتریان خود بهترین کیفیت را ارائه میدهیم. محصولات ما از بهترین مزارع زعفران در روستای فندخت زیرکوه قائنات تامین میشوند و با استفاده از تکنولوژیهای پیشرفته و فرآیندهای بهینهسازی فرآوری، به شما ارائه میگردند.